Computer Aided Design & Drafting CADD Users Manual Caltrans
Table Of Content
- Computational Approaches for Drug Target Identification
- Hybrid computational approaches
- Computer-Aided Drug Design Methods
- Modular synthon-based approaches
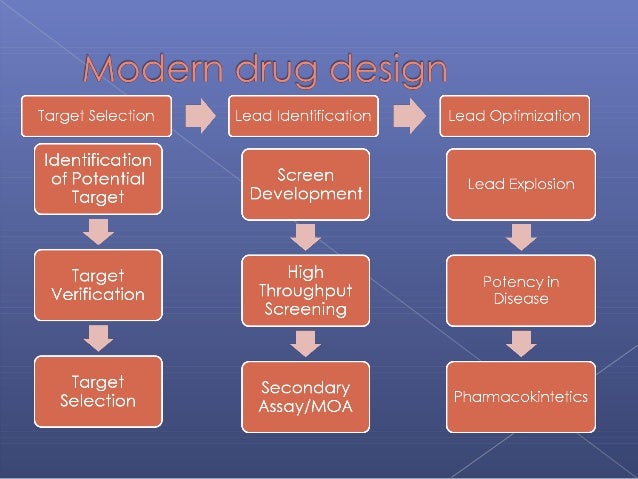
- How a computer can help in drug design and discovery?
- Computer Aided Drug Design and its Application to the Development of Potential Drugs for Neurodegenerative Disorders
The CADD methods presented in the chapter such as SILCS for SBDD or CSP for LBDD take this issue into account and thus have advantages over other CADD methods that only rely on single crystal structure or limited ligand conformations. CADD can be separated into ligand or hit identification and ligand or hit optimization, with both SBDD and LBDD methods useful in the appropriate context. Database screening methods are often used for hit identification (59) while a number of methods may be used for hit optimization (4, 24, 60). Below we present a collection of methods that may be used for both ligand identification and optimization.
Computational Approaches for Drug Target Identification
For examples, Trylska et al. studied the effects of mutations at the bacterial ribosomal A-site using molecular dynamics (MD) simulations to reveal the origins of bacterial resistance to aminoglycosidic antibiotics (5). He has contributed to the development of a number of technologies in this area, including the creation and maintenance of BindingDB, the first publicly accessible database of protein-small molecule binding data. Dr. Gilson is currently on the faculty of UC San Diego’s Skaggs School of Pharmacy and Pharmaceutical Sciences. The most reliable and accurate region in the 3D structure in the protein-ligand complex is the analysis and prediction of the region in a protein where ligand can occupy its best pose.
Hybrid computational approaches
RNA-dependent RNA polymerase (RdRp) is the cleavage product of the polyproteins 1a and 1ab from ORF1a and ORF1ab and is involved in the replication and transcription of the SARS-CoV-2 genome [120]. The catalytic core of the enzyme resembles the human right hand with differentiated palm, fingers, and thumb domains. Targeting this enzyme to halt the viral replication seems an effective therapeutic approach since the active site of the RdRp is a highly conserved and accessible region [121]. Nsp15 is a uridine-specific endoribonuclease involved in RNA processing and widely distributed in all kingdoms of life.
Computer-Aided Drug Design Methods
The performance and generalizability of ML and DL methods for these and other targets remain to be tested. Although the impacts of the recent structural revolution17 and computing hardware in drug discovery28 are comprehensively reviewed elsewhere, here we focus on the ongoing expansion of accessible drug-like chemical spaces as well as current developments in computational methods for ligand discovery and optimization. We detail how emerging computational tools applied in gigaspace can facilitate the cost-effective discovery of hundreds or even thousands of highly diverse, potent, target-selective and drug-like ligands for a desired target, and put them in the context of experimental approaches (Table 1). Although the full impact of new computational technologies is only starting to affect clinical development, we suggest that their synergistic combination with experimental testing and validation in the drug discovery ecosystem can markedly improve its efficiency in producing better therapeutics. Previously, we published a chapter in the first edition of this book that was dedicated to an overview of CADD and included information on routinely utilized protocols, especially tools used in our laborotary, towards the design of antibotic theraputics (4).

BMC Chemistry
Computer-Aided Drug Design (CADD) strategies have become indispensable tools in modern drug discovery and development. Besides academia, large and small-sized pharmaceutical and biotechnology companies have been using intelligent software to assist in the discovery or optimization of bioactive compounds. This includes, but is not limited to, Aliskiren, Boceprevir, Captopril, Dorzolamide, Nolatrexed, Oseltamivir, Rupintrivir, Saquinavir and Zanamivir [2].
In order to achieve this, molecular docking tools are used to generate a set of different ligand binding poses and a scoring function is used to estimate the binding affinities of generated poses to identify the best binding mode. The energy change caused by ligand/receptor complex formation, is given by the Gibbs free energy (ΔG) and the binding constant (Kd) [49, 50]. The binding energy of a complex is predicted by evaluating physicochemical features involved in ligand-receptor binding, which include desolvation, intermolecular interactions, and entropic effects [51]. Sehgal et al. identified a number of compounds active against HSPB8 based on molecular docking results [52].
This review article provides useful insights into some of the common in silico methods used in CADD and how these methods have been currently used and can be of help in the drug discovery process of COVID-19. Molecular docking is a computational process widely used for rapidly predicting the binding modes and affinities of small molecules against their target molecules (usually proteins) [35, 36]. This in silico process has achieved a position of great importance in the drug discovery field [21, 36-38]. Molecular docking has emerged over the last two decades and is now considered an indispensable tool for CADD and in the structural biology field, and has been shown to be more efficient than traditional drug discovery methods.
The simulation results provide clues for the design of new membrane disruptors to treat mcr-1 infections. The development of broadly generalizable or even universal models is the key aspiration of AI-driven drug discovery. One of the directions here is to extract general models of binding affinities (binding score functions) from data on both known ligand activities and corresponding protein–ligand 3D structures, for example, collected in the PDBbind database81 or obtained from docking. Such models explore various approaches to represent the data and network architectures, including spatial graph-convolutional models82,83, 3D deep convolutional neural networks84,85 or their combinations86.

Both ligand-based and structure-based drug design approaches have been widely used in the drug discovery process against coronavirus disease-19 (COVID-19), an infectious viral disease caused by SARS-CoV-2. To date, only a few drug-candidate molecules have undergone clinical trials, and these molecules are mostly repurposed approved drugs (Figure 2). With the ever-increasing demand for drugs, the selection of biologically active structures draws greater attention. The knowledge of structural biology and the power of computers has become possible to exploit the use of computational methods to identify active libraries to resolve the molecular obesity problem. Docking is a useful CADD tool to predict binding orientation of a ligand molecule within target binding site as well as to evaluate its binding strength (70). Traditional docking methods only consider rigid or limited protein flexibility and ignore or treat the contribution of desolvation to binding in an empirical way.
These methods, including MolPal25, Active Learning110 and DeepDocking111, report as much as 14–100 reduction in the computational cost for libraries of 1.4 billion compounds, although it is not clear how they would scale to rapidly growing chemical spaces. In virtual screening approaches, a synergetic use of physics-based docking with data-based scoring functions may be highly beneficial. Moreover, if the physics-based and data-based scoring functions are relatively independent and both generate enrichment in the selected focused libraries, their combination can reduce the false-positive rates and improve the quality of the hits.
First Ever AI Solution to Integrate Drug Discovery and Synthesis - Lab Manager Magazine
First Ever AI Solution to Integrate Drug Discovery and Synthesis.
Posted: Tue, 02 Jan 2024 08:00:00 GMT [source]
While FEP and molecular mechanics (MM) with Poisson–Boltzmann (PB) and surface area solvation (MM/PBSA) as well as MM/generalized-Born SA (GBSA) methods (107) do account desolvation, these are computational demanding approaches that limits their utility in CADD. A novel method designed to overcome this drawback is the SILCS-MC (19) docking method put forward by our laboratory. This takes advantage of the use of GCMC/MD simulations of the protein in aqueous solution with selected organic solutes to precompute the GFE FragMaps that are free energy functional group affinity patterns that encompass the entire protein and account for protein flexibility and desolvation contributions (108–110). SILCS-MC then involves simply assigning the GFE value for the appropriate FragMap type to each atom in the molecule and summing those values to get the LGFE score. MC conformational sampling is then performed to allow the orientation and conformation of the ligands to relax in the field of the GFE FragMaps. This allows for SILCS-MC docking to be performed in a highly computationally efficient fashion while achieving a level of accuracy similar to highly expensive FEP methods (109).
Melanie Cocco has a PhD in Organic Chemistry from Penn State and was an NIH postdoctoral fellow at Yale in the Department of Biophysics and Biochemistry. The Cocco lab at UCI uses Nuclear Magnetic Resonance (NMR) spectroscopy as well as other biophysical and molecular biology techniques to study membrane proteins and DNA binding proteins. The UC DDC is governed by a diverse group of experts in drug discovery and development that serve as site lead representatives for their respective UC campuses. The basic idea is that the overall binding free energy can be decomposed into independent components that are known to be important for the binding process. Each component reflects a certain kind of free energy alteration during the binding process between a ligand and its target receptor.



Comments
Post a Comment